Heather Kopecky1, William Maier MD, Diman Lamichhane MD
PNWMSRJ. Published online Sept 25th, 2020.Abstract
Introduction & Objective: Anti-MDA5 positive dermatomyositis does not contain the same typical skin findings and progressive muscle weakness as classical dermatomyositis and can be difficult to diagnose and treat. The objective of this paper is to highlight the rapidly progressive onset of necrotic skin lesions in a patient with cutaneous disease refractory to initial treatments. Case Presentation: 49 yo Vietnamese male presented with clinically non-specific skin rash and isolated urine protoporphyrins leading to initial diagnosis of porphyria cutanea tarda. After lack of response to prednisone with worsening and transforming skin rash and proximal muscle weakness, a further workup was initiated and ultimately revealed anti-MDA5 positive dermatomyositis with refractory skin disease and without rapidly progressive interstitial lung disease. The patient has remained stable on a combination of IVIG, rituximab, wound care, and Bactrim prophylaxis. Conclusion: The quick diagnosis and aggressive treatment of anti-MDA5 positive dermatomyositis is necessary to treat painful and rapidly progressive skin ulcerations. A multidisciplinary approach involving rheumatology, dermatology, pulmonology, pathology, radiology, and infectious disease is paramount to the successful treatment and improved quality of life for these patients.
Introduction
Dermatomyositis is a multisystem, idiopathic, inflammatory myopathy. The incidence is approximately 8 cases/million population/year with a bimodal age distribution with both juvenile and adult phenotypes.1 Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis, first identified in 2005,2 is a subtype of dermatomyositis presenting with clinically distinct skin and mucous membrane findings. MDA5 is a pattern recognition receptor capable of detecting double and single-stranded RNA, namely in viruses. Activation of MDA5 causes downstream transcription of type I interferons. The clinical presentation of anti-MDA5 dermatomyositis differs from that of typical cutaneous dermatomyositis and the diagnosis is often overlooked. This is important because patients with this disease process have a significant risk for developing rapid interstitial lung disease (ILD) and a delay in diagnosis can be devastating.3,4
Cutaneous manifestations of classic dermatomyositis include heliotrope rash and Gottron’s papules with malar erythema, photo-distributed poikiloderma, violaceous erythema, periungual and cuticular changes, and alopecia.5 In contrast, anti-MDA5 dermatomyositis tends to present with distinct mucocutaneous findings. Large cutaneous ulcers develop in 82% of cases and usually appear as deep ulcerations with necrotic centers and a hyperkeratotic crust. Generally, these ulcers develop overlying the usually very recognizable Gottron’s papules and on the elbows and knees. Additionally, palmar papules and gum pain are frequently noted. 3 A study conducted at Stanford University agrees with previous studies showing that in patients with anti-MDA5 dermatomyositis, skin disease predominates with absent or very mild muscle disease. Additionally, this cutaneous necrosis is very challenging to treat and leads to significant quality of life impairment (even more so than psoriasis and atopic dermatitis).6,7 While the exact pathophysiology of cutaneous disease in dermatomyositis is still unknown, it is thought that there is an increase in cutaneous vasculopathy compared to the muscular vasculopathy of classic dermatomyositis. This is possibly related to the overexpression of MDA5 in individuals with this phenotype, as the MDA5 leads to release of type I interferons which have vasculopathic effects of their own.7
Case Presentation:
A 49-year-old Vietnamese male with a medical history complicated by alpha thalassemia, alcoholism (sober since June 2018), and chronic hepatitis B presented 5 days post left carpal tunnel release procedure with a presumed left septic wrist and bacteremia. During hospitalization, the patient developed a diffuse violaceous rash of the bilateral hands, arms, and upper chest. Lab studies revealed elevated urine porphyrins leading to a diagnosis of porphyria cutanea tarda and treatment with a course of high dose prednisone. One week post discharge for bacteremia, the patient returned with concerns of hypotension, proximal muscle weakness, and worsening skin rash on the bilateral dorsal hands. Lab workup revealed normal CK levels but aldolase elevated to 11.8 U/L and ferritin levels of 2700 ng/mL. Physical description of the worsening skin rash showed pink thin papules to plaques on the PIP and MCPs of bilateral hands and ill-defined erythematous scaly thin plaques on the extensor arms, upper chest, and upper back. A skin biopsy revealed vacuolar dermatitis with plasma cells which the pathologist commented may be consistent with dermatomyositis but is not suggestive of porphyria cutanea tarda. A muscle biopsy of the R deltoid performed at this hospitalization revealed degenerating/regenerating myofibers, a sparse macrophage infiltrate, and no increase in lymphocytic inflammation. The pathologist commented that the only features supporting a diagnosis of dermatomyositis included some slight perifascicular atrophy but were rather non-specific. The autoantibody assay returned positive for anti-MDA5 antibodies supporting the diagnosis of dermatomyositis presenting with worsening skin rash and minor proximal muscle weakness. Chest CT revealed scattered bilateral ground glass opacities and the patient was started on 60 mg prednisone and mycophenolate mofetil 1000mg bid.
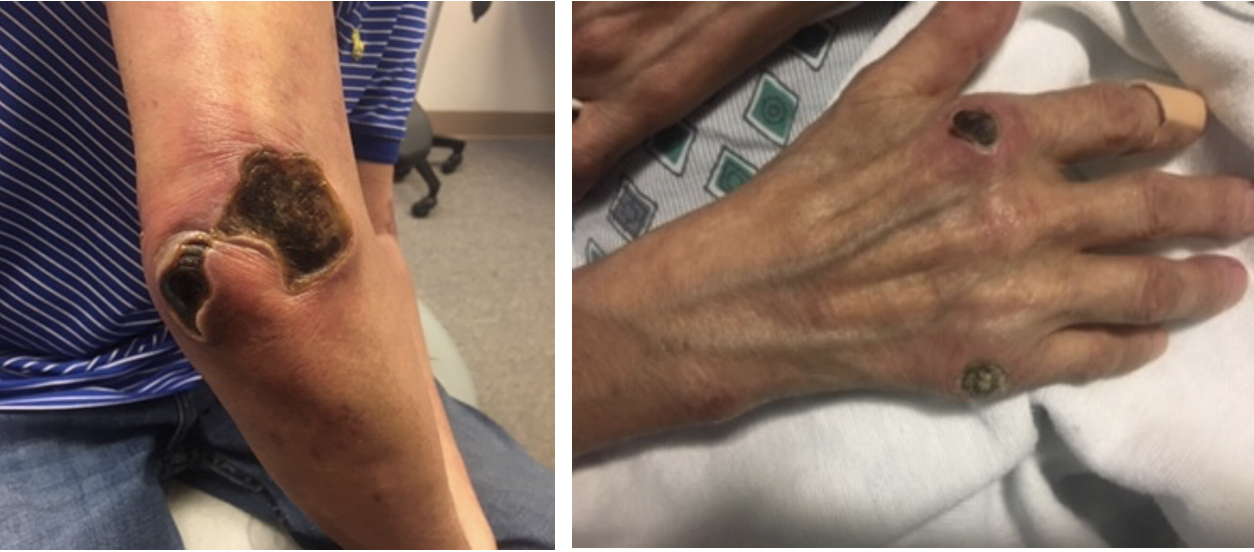
Three weeks after starting prednisone and mycophenolate mofetil, the patient again presented to the emergency department for worsening skin rash and confusion. Labs showed worsening transaminitis, thrombocytopenia, and chest CT with bilateral ground glass opacities. The patient was admitted, the mycophenolate mofetil was discontinued due to concerns of liver toxicity and worsening symptoms, and the patient was started on IV solumedrol 500 mg daily for 3 days and IVIG 2 gm/kg given over 3 days. At this time, the patient was also switched to rituximab 1000mg. The previously pink, papular rash now appeared as punched out ulcerations with overlying eschars on the bilateral elbows, posterior shoulders, bilateral dorsal feet, right 5th MCP, right index MCP and bilateral pinna (Figure 1). The eschars on the right shoulder and right elbow were removed with sharp dissection but the others were too adherent. He was started on collagenase and recommended weekly wound care visits and 10 mg daily prednisone on discharge.
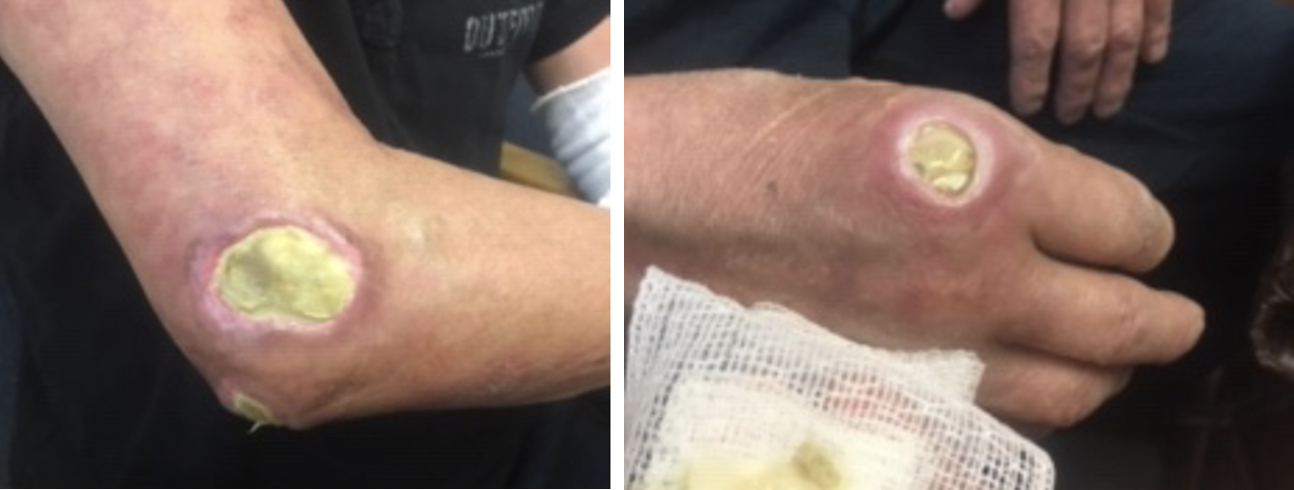
Follow up two months after starting rituximab 1000mg infusions, 10 mg daily prednisone, 3 times weekly IVIG, as well as careful wound care showed slowly improving skin lesions (Figure 2) without development of new lesions, stable ILD as noted by repeat CT scan, and improved clinical complaints of muscle weakness. The patient was treated twice for pseudomonas pneumonia but recovered well. Clinically, the patient reported improved skin lesions and no breathing concerns. Collaboration between rheumatology and dermatology agreed on continuation of the rituximab 1000mg 2 doses every 6 months, IVIG as the ulcers continue to improve, 5 mg prednisone daily, and Bactrim 3 times weekly for bacterial prophylaxis. The patient is doing well.
Discussion:
Anti-MDA5 dermatomyositis can pose a diagnostic challenge on initial presentation. Interestingly, it seems that in this patient the trigger for the clinical development of dermatomyositis was the septic wrist. There was a 7 week delay in reaching the diagnosis of anti-MDA5 dermatomyositis as the elevated urine porphyrins were the only real diagnostic clue for the somewhat non-specific initial skin rash. The rash began as diffuse and violaceous, transformed into pink and papular, then finally turned to deep ulcerations. It wasn’t until the patient began developing rapidly worsening skin lesions prompting biopsy and proximal muscle weakness that the definitive diagnosis was reached. This patient is fortunate that he has been spared from the rapidly progressive ILD that often comes with this subtype of disease. The estimated prevalence of ILD in Asian populations with anti-MDA5 dermatomyositis is 92%-100%.3 He has, however, struggled with recurrent pulmonary infections.
Aggressive treatment for this patient’s anti-MDA5 dermatomyositis was warranted especially given his very high ferritin values which carries a strong association with disease activity in these patients. According to Kurtzman et al, elevated serum ferritin levels (≥500 ng/mL) suggests decreased survival and the strength of this association increases with increasing ferritin levels (≥1600 ng/mL).3 The decision to begin treatment with mycophenolate mofetil in this patient is consistent with the first line steroid-sparing agent used successfully in other case studies, however this patient did not respond favorably and was switched to rituximab plus IVIG with beneficial result and skin improvement. This treatment combination of rituximab plus IVIG is consistent with new research demonstrating favorable outcomes in patients with refractory autoimmune processes, especially those presenting cutaneously.8
The multidisciplinary team of rheumatologists, dermatologists, pathologists, radiologists, infectious disease specialists, and pulmonologists who evaluated this patient ultimately reached the correct diagnosis and a suitable treatment plan relatively quickly. Had this patient had to wait longer for aggressive treatment, it is likely given his high disease markers, quickly progressive skin findings, and his propensity for sepsis that morbidity would have been greatly increased.
Learning Points:
1) Anti-MDA5 dermatomyositis can be difficult to diagnose given the lack of myopathy and the different mucocutaneous presentation compared to classical dermatomyositis
2) Early recognition of the associated skin lesions is important given the high morbidity from rapidly progressive ILD. Ferritin and aldolase are useful disease markers.
Acknowledgments:
I would like to acknowledge doctors William Maier and Diman Lamichhane for presenting me with this case for review. They are both wonderful student mentors and teachers. Dr. Maier and Dr. Lamichhane are practicing rheumatologists in Eugene, OR.
References:
- Milisenda J, Doti P, Prieto-Gonzalez S, et al. Dermatomyositis presenting with severe subcutaneous edema: Five additional cases and review of the literature. Seminars in Arthritis and Rheumatism. 2014; 44:228-233. https://pubmed.ncbi.nlm.nih.gov/24830790/.
- Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinical amyopathic dermatomyositis. Arthritis Rheumatology. 2005; 52(5):1571-1576. https://pubmed.ncbi.nlm.nih.gov/15880816/.
- Kurtzman D, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: A concise review with an emphasis on distinctive clinical features. Journal of the American Academy of Dermatology. 2018; 78(4): 776-785. https://pubmed.ncbi.nlm.nih.gov/29229575/.
- Callen J. Cutaneous manifestations of dermatomyositis and their management. Current Rheumatology Reports. 2010; 12: 192-197. https://pubmed.ncbi.nlm.nih.gov/20425525/.
- Callen J, Wortmann R. Dermatomyositis. Clinics in Dermatology. 2006; 24:363-373. https://pubmed.ncbi.nlm.nih.gov/16966018/.
- Hundley JL, Carroll CL, Lang W, et al. Cutaneous symptoms of dermatomyositis significantly impact patients’ quality of life. Journal of the American Academy of Dermatology. 2006; 54:217. https://pubmed.ncbi.nlm.nih.gov/16443050/.
- Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): A retrospective study. Journal of the American Academy of Dermatology. 2011; 65(1):25-34. https://pubmed.ncbi.nlm.nih.gov/21531040/.
- Ahmed A.R., Kaveri S. Reversing autoimmunity combination of rituximab and intravenous immunoglobulin. Frontiers in Immunology. 2018; 9: 1189. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6058053/.
Article information:
Published Online: Sept 25th, 2020.
IRB Approval: This study did not require IRB approval
Conflict of Interest Declaration: All Authors have no conflicts of interest to disclose
Funding Source/Disclosure: No funding was solicited or granted for this research project. All researchers volunteered their time